





In agosto, l’associazione Public Health and Medical Professionals for Transparency (PHMPT) ha presentato una richiesta alla Food and Drug Administration (FDA) statunitense in base al Freedom of Information Act (FOIA) per avere tutti i dati contenuti nel dossier da prodotto biofarmacologico del vaccino COVID-19 della Pfizer – un corpo di informazioni che comprende circa 329.000 pagine.
La FDA, sostenendo che il suo Centro per la valutazione e la ricerca dei prodotti biofarmacologici, per carenza di personale, non aveva la capacità di espungere rapidamente i contenuti legalmente esenti, come le informazioni di proprietà della Pfizer e le informazioni private personali dei partecipanti alla sperimentazione, l’agenzia ha chiesto di poter rilasciare solo 500 pagine di questi dati al mese, rendendo così necessari 55 anni per la divulgazione completa.
L’agenzia ha poi richiesto fino a 75 anni per completare il compito. Al 17 novembre, era stata rilasciata solo una piccolissima parte dei dati in questione.
Qui tratterò uno di questi documenti rilasciati, l’ “Analisi cumulativa dei rapporti sugli eventi avversi post-autorizzazione“. [La redazione di] questo documento costituisce parte della responsabilità della Pfizer per la farmacovigilanza nell’ambito del suo Accordo per la licenza biologica (Biological License Agreement, BLA) con la FDA.
Con farmacovigilanza si intende la scienza e le attività relative al rilevamento, alla valutazione, alla comprensione e alla prevenzione degli effetti avversi o di qualsiasi altro problema legato ai farmaci.
Prima di esaminare la quantità, la gravità e la natura degli eventi avversi inclusi in questo documento, vale la pena soffermarsi a considerare quanto questo rapporto avrebbe dovuto essere significativo per il pubblico.
Il vaccino Pfizer non aveva ancora completato i test di sicurezza ed efficacia, eppure questo prodotto veniva rapidamente somministrato a una popolazione sana molto più grande del gruppo della sperimentazione clinica del vaccino.
La FDA e la Pfizer erano ben consapevoli che rischi molto concreti, se esistevano, non avrebbero potuto essere identificati in base alle sole sperimentazioni. Non c’erano abbastanza partecipanti e i partecipanti non erano stati osservati per molto tempo.
Tutto può sembrare a posto se si fa un esperimento su 20.000 persone, ma cosa succede quando si sperimenta lo stesso farmaco su un milione di persone?
L'”Analisi cumulativa dei rapporti sugli eventi avversi post-autorizzazione” avrebbe dovuto essere la rassicurazione del tipo “finora tutto a posto” cercata dalla FDA. Perché è stato necessario spingere la FDA a pubblicare queste informazioni tramite un’ordinanza del tribunale?
Nella sezione argomentativa del documento (sezione 4), la Pfizer assicura alla FDA che “… esegue frequenti e rigorosi rilevamenti dei segnali sui casi BNT162b2”.
Cosa significa rilevamento “rigoroso” dei segnali? Forse che la Pfizer ha intervistato un gran numero di destinatari del vaccino per trovare eventi, che ha poi indagato? No.
Questa relazione è semplicemente una compilazione di segnalazioni non richieste, in altre parole passive, di eventi avversi portati direttamente all’attenzione della Pfizer dai vaccinati, casi segnalati dalle autorità sanitarie, casi pubblicati nella letteratura medica, casi provenienti da programmi di marketing sponsorizzati dalla Pfizer, studi non interventistici e casi di eventi avversi gravi riportati da studi clinici indipendentemente da una valutazione di causalità.
Nella relazione, la Pfizer ammetteva che “non si conosce l’entità della sottosegnalazione”.
È dato per scontato che la segnalazione passiva porterà ineluttabilmente a una sottosegnalazione. Tuttavia, secondo il rapporto della Pfizer:
“A causa del gran numero di segnalazioni di eventi avversi spontanei ricevuti per il prodotto, il MAH (Marketing Authorisation Holder) ha dato la priorità ai casi gravi, al fine di soddisfare le scadenze regolamentari accelerate di segnalazione e garantire che queste segnalazioni siano disponibili per l’attività di rilevamento e valutazione dei segnali”.
Gli autori continuavano:
“La Pfizer ha anche preso una [sic] azioni multiple per contribuire ad affrontare il grande aumento di segnalazioni di eventi avversi. Questo include significativi miglioramenti tecnologici, [sic] e soluzioni di processo e di flusso di lavoro, così come l’aumento del numero del personale dedito all’inserimento dati e all’elaborazione dei casi”.
In altre parole, il numero di eventi avversi riportati superava le aspettative della Pfizer, eppure il produttore del vaccino concludeva: “I risultati di queste analisi di rilevamento dei segnali sono coerenti con il profilo di sicurezza noto del vaccino”.
Questa affermazione paradossale si rivelerà un indizio importante mentre dissezioniamo i dati qui sotto.
Cosa rivela il documento?
Fino al 28 febbraio, un totale di 42.086 destinatari (casi) ha riportato 158.893 eventi, o reazioni avverse (AE) al vaccino Pfizer. Circa il 50% di questi eventi era considerato grave.
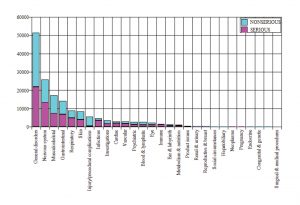
Una panoramica delle caratteristiche dei destinatari è data qui:
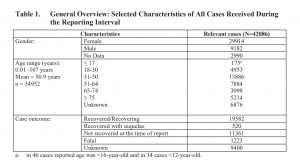
Da notare che 1.223 destinatari del vaccino hanno avuto un esito fatale. Più di 11.000 non si erano ripresi. L’esito per 9.400 casi era sconosciuto. Quasi tre quarti erano donne.
Questi numeri sono preoccupanti, ma rappresentano una preoccupazione significativa per la sicurezza? La risposta a questa domanda dipende interamente dal numero di persone che erano state vaccinate fino a quel momento.
Pfizer ha fornito questo numero alla FDA nella sezione generale del documento, sezione 3.1.1. – ma nel documento rilasciato sotto la richiesta FOIA, quel numero è stato occultato:
“Si stima che circa (b) (4) dosi di BNT162b2 siano state spedite in tutto il mondo a partire dal ricevimento della prima autorizzazione temporanea per la fornitura di emergenza tra il 1 dicembre 2020 e il 28 febbraio 2021.”
Nel passo sopra, “(b)(4)” indica che questo numero è stato censurato.
Il numero cumulativo di dosi distribuite in tutto il mondo al 28 febbraio non è un’informazione proprietaria, né costituisce dati personali e privati di individui.
Eppure senza questo numero chiave non c’è modo di calcolare l’incidenza degli eventi gravi, il che è un segnale di sicurezza.
La FDA ha scelto, senza alcuna spiegazione o giustificazione legale, di non rivelare questo dato cruciale.
Nonostante l’evidente intenzione della FDA di offuscare, la Pfizer fornisce un mezzo per stimare questo numero concludendo inequivocabilmente: “… queste analisi di rilevamento dei segnali sono coerenti con il profilo di sicurezza noto del vaccino”.
Qual era il profilo di sicurezza noto del vaccino?
Al 28 febbraio, l’unico profilo di sicurezza noto del vaccino era determinato dai risultati iniziali degli studi di fase 3 dell’autunno 2020.
Su 21.621 destinatari del vaccino Pfizer, 126 [Polack FP, Thomas SJ, Kitchin N, et al., NEJM, Tabella S3] subirono un evento avverso grave nelle sperimentazioni. Questo è all’incirca un evento avverso grave su 171,6 destinatari.
Quindi, se questi dati sono coerenti con il profilo di sicurezza noto, e fino a quel momento si erano verificati circa 79.000 eventi avversi gravi, possiamo stimare che erano state distribuite circa 13.550.000 (79.000 x 171,6) dosi.
Certamente c’è incertezza in questo calcolo. Forse era implicita una diversa interpretazione del profilo di sicurezza.
Tuttavia, la Pfizer ha riportato il numero di dosi che erano state distribuite, non somministrate.
Sarebbero state somministrate meno dosi di quelle consegnate. Inoltre, gli eventi avversi gravi nelle sperimentazioni sono stati distribuiti tra i partecipanti che erano completamente vaccinati (avendo ricevuto due dosi).
Qui stiamo usando il numero di dosi come denominatore. Questa stima rientrerà nel limite inferiore della vera incidenza degli eventi avversi.
In altre parole, utilizzando queste ipotesi stiamo concedendo al vaccino della Pfizer il massimo beneficio del dubbio.
Usando questa stima delle dosi totali somministrate, l’incidenza di un esito fatale è 1223/13,55 milioni o 1 su 11.079.
Sequele permanenti (condizioni che risultano come conseguenza della vaccinazione) = 520/13,55 milioni, o 1 su 26.057. Inoltre, 11.361 su 13,55 milioni, o 1 su 1.193, non si erano ancora ripresi da un evento avverso.
La Pfizer ha inspiegabilmente scelto di raggruppare i destinatari che “si sono ripresi” con quelli che si stavano “riprendendo”. Quanti di questo grande gruppo (19.582) soffrivano ancora di danni al momento del rapporto? Su quali basi la Pfizer ha stabilito che un ricevente aveva ancora una possibilità di recupero completo?
Senza alcun chiarimento da parte del produttore del vaccino, siamo costretti a metterli insieme a un altro grande gruppo di 9.400 il cui risultato finale era “sconosciuto” – lasciandoci con un limite elevato di 1 su 466 destinatari che hanno avuto un risultato indeterminato.
Anche se non è stato dimostrato che nessuno di questi eventi avversi e decessi fosse direttamente o indirettamente causato dalla vaccinazione, la Pfizer ha offerto ulteriori dati preoccupanti riguardo agli eventi avversi di “particolare interesse” (AESI).
Secondo la Pfizer si sono verificati 1.403 AESI cardiovascolari, 932 ematologici, 3.600 muscoloscheletrici, 501 neurologici e 3.674 “altri” AESI gravi, tutti con un tempo mediano di insorgenza di 24 ore o meno dalla vaccinazione.
I 275 ictus e i 449 casi di paralisi facciale riportati si sono verificati con un tempo mediano di insorgenza di due giorni dalla vaccinazione.
Sebbene sia impossibile stabilire un nesso causale inattaccabile tra la vaccinazione e il danno in questo momento, la relazione temporale tra loro è correlativa e altamente suggestiva di causalità.
Tuttavia, gli autori del rapporto della Pfizer concludevano alla fine di ogni categoria AESI che “Questa revisione cumulativa dei casi non solleva nuove questioni di sicurezza”.
Il rapporto includeva anche 24 casi gravi in bambini di meno di 12 anni. Di questi, 13 casi non si erano ancora risolti al momento della segnalazione. L’età media di questi destinatari era di 3,7 anni.
Dobbiamo supporre che pochissimi bambini di quell’età siano stati inoculati a quel tempo, dato che la Pfizer aveva l’autorizzazione per l’uso solo sugli adulti. Dato che il numero di bambini vaccinati non è riportato, non possiamo sapere quale sia il rischio di lesioni nei bambini sotto i 12 anni.
Conclusioni
Le ripetute assicurazioni della Pfizer che non esistono nuovi problemi di sicurezza sono insincere nel migliore dei casi.
La FDA è stata apertamente ostruzionista nel non rivelare informazioni cruciali per poter fare una valutazione accurata dei danni. Tuttavia, usando stime ragionevoli basate sulle affermazioni della stessa Pfizer e sui dati delle sperimentazioni pubblicati, è probabile che un segnale di sicurezza esista – e che questo segnale di sicurezza sia stato ignorato proprio dall’organizzazione che dovrebbe cercarlo: la FDA.
L’incidenza stimata dalla Pfizer per la potenziale fatalità del vaccino, 1 su 11.079, è circa il doppio di quella riportata in VAERS. Dato che le potenziali vittime del vaccino in questo documento sono state riportate passivamente, possiamo supporre che l’incidenza reale sia più alta.
Analisi più complete hanno dimostrato un fattore di sottosegnalazione al VAERS di mortalità da vaccino che si avvicina a 41 o più.
Sottosegnalazioni a parte, la vera e crescente tragedia è che fino a quando non si è dimostrato che un danno associato alla vaccinazione è causato da essa, il danno rimane, a tutti gli effetti, un segnale inesistente proprio per le istituzioni responsabili della salute e della sicurezza pubblica.
Su quali basi noi, come medici e fornitori di assistenza sanitaria, possiamo assicurare ai nostri pazienti che questo vaccino è sicuro se gli eventi avversi non vengono investigati o addirittura riconosciuti?
Un assenso della FDA è davvero sufficiente?
Non dovremmo piuttosto chiedere trasparenza, discussioni o almeno dati non censurati? Cosa si aspetta il pubblico da noi?