





En août, l’organisation Public Health and Medical Professionals for Transparency (PHMPT) a soumis une demande en vertu de la loi sur la liberté d’information (FOIA) à la Food and Drug Administration (FDA) pour obtenir toutes les données contenues dans le dossier du produit biologique du vaccin contre la COVID-19 de Pfizer – un ensemble d’informations comprenant quelque 329 000 pages.
La FDA, disputant que son Center for Biologics Evaluation and Research dispose d’un personnel insuffisant et n’a pas la capacité d’expurger rapidement les documents exemptés par la loi, tels que les informations exclusives de Pfizer et les informations privées des participants aux essais, l’agence a demandé l’autorisation de ne publier que 500 pages de ces données par mois, ce qui nécessite 55 ans pour une divulgation complète.
L’agence a ensuite demandé jusqu’à 75 ans pour mener à bien cette tâche. Au 17 novembre, seule une partie des données en question avait été publiée.
Je discuterai ici de l’un de ces documents publiés, l’ « analyse cumulative des rapports d’événements indésirables post-autorisation » . Ce document constitue une partie de la responsabilité de Pfizer en matière de pharmacovigilance dans le cadre de son accord de licence biologique avec la FDA.
La pharmacovigilance désigne la science et les activités relatives à la détection, l’évaluation, la compréhension et la prévention des effets indésirables ou de tout autre problème lié aux médicaments.
Avant d’examiner la quantité, la gravité et la nature des événements indésirables inclus dans ce document, il convient de faire une pause et de se demander quelle importance ce rapport aurait dû avoir pour le public.
Le vaccin de Pfizer n’avait pas encore achevé tous les tests de sécurité et d’efficacité, mais son produit était rapidement déployé sur une population saine dont la taille dépassait celle de l’essai clinique du vaccin.
La FDA et Pfizer étaient parfaitement conscients que des risques très réels, s’ils existaient, n’auraient pas pu être identifiés à partir des seuls essais. Il n’y avait pas assez de participants, et les participants n’avaient pas été observés pendant très longtemps.
Tout peut sembler correct si vous expérimentez sur 20 000 personnes, mais que se passe-t-il lorsque vous expérimentez sur un million de personnes ?
L’ « analyse cumulative des rapports d’événements indésirables post-autorisation » aurait dû être la garantie que « tout semble bon jusqu’à présent » que la FDA recherchait. Pourquoi a-t-il été nécessaire de contraindre la FDA à rendre ces informations publiques par une décision de justice ?
Dans la section discussion du document (section 4), Pfizer assure à la FDA qu’il « … effectue une détection fréquente et rigoureuse des signaux sur les cas de BNT162b2 » .
Que signifie la détection « rigoureuse » des signaux ? Pfizer a-t-elle enquêté auprès d’un grand nombre de personnes ayant reçu le vaccin pour détecter les effets indésirables et les étudier ? Non, ce n’est pas le cas.
Ce rapport n’est qu’une compilation de rapports non sollicités, c’est-à-dire passifs, d’événements indésirables directement portés à l’attention de Pfizer par des destinataires, de cas signalés par les autorités sanitaires, de cas publiés dans la littérature médicale, de cas issus de programmes de marketing parrainés par Pfizer, d’études non interventionnelles et de cas d’événements indésirables graves signalés par des études cliniques, indépendamment de l’évaluation du lien de causalité.
Dans le rapport, Pfizer a admis que « l’ampleur de la sous-déclaration est inconnue » .
Il est largement admis que la déclaration passive conduira inéluctablement à une sous-déclaration. Néanmoins, selon le rapport de Pfizer :
« En raison du grand nombre de déclarations spontanées d’effets indésirables reçues pour le produit, le titulaire de l’autorisation de mise sur le marché (MAH) a donné la priorité au traitement des cas graves, afin de respecter les délais de déclaration réglementaire accélérés et de s’assurer que ces déclarations sont disponibles pour l’activité de détection et d’évaluation des signaux. »
Les auteurs poursuivent :
« Pfizer a également pris de multiples mesures à l’adresse [sic] pour atténuer la forte augmentation des rapports d’effets indésirables. Cela comprend des améliorations technologiques importantes, [sic] et des solutions en matière de processus et de flux de travail, ainsi que l’augmentation du nombre de collègues chargés de la saisie des données et du traitement des dossiers. »
En d’autres termes, le nombre d’événements indésirables signalés a dépassé les attentes de Pfizer, et pourtant le fabricant du vaccin a conclu : « Les résultats de ces analyses de détection des signaux sont conformes au profil de sécurité connu du vaccin. »
Cette déclaration paradoxale s’avérera être un indice important lorsque nous disséquerons les données ci-dessous.
Que révèle le document ?
Jusqu’au 28 février, un total de 42 086 receveurs (cas) ont signalé 158 893 événements, ou réactions indésirables au vaccin de Pfizer. Environ 50 % de ces événements ont été jugés graves.
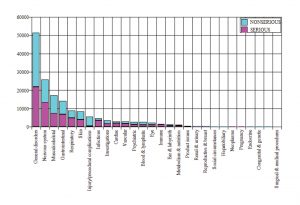
Un aperçu des caractéristiques des bénéficiaires est donné ici :
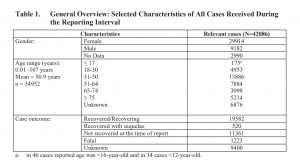
Il convient de noter que 1 223 personnes ayant reçu le vaccin ont connu une issue fatale. Plus de 11 000 d’entre eux ne s’étaient pas rétablis. Le résultat de 9 400 personnes était inconnu. Près des trois quarts étaient des femmes.
Ces chiffres sont préoccupants, mais représentent-ils un problème de sécurité important ? La réponse à cette question dépend entièrement du nombre de personnes qui ont été vaccinées jusqu’à ce moment-là.
Pfizer a fourni ce numéro à la FDA dans la section de l’aperçu général du document, section 3.1.1. – mais dans le document publié dans le cadre de la demande de liberté d’information, ce nombre a été expurgé :
« On estime qu’environ (b) (4) doses de BNT162b2 ont été expédiées dans le monde entier entre la réception de la première autorisation temporaire d’approvisionnement d’urgence du 01 décembre 2020 jusqu’au 28 février 2021. »
Dans le texte ci-dessus, « (b) (4) » indique que ce numéro a été expurgé.
Le nombre cumulé de doses distribuées dans le monde au 28 février n’est pas une information exclusive et ne constitue pas une donnée personnelle et privée des individus.
Or, sans ce chiffre clé, il est impossible de calculer l’incidence des événements graves, c’est-à-dire un signal de sécurité.
La FDA a choisi, sans explication ni justification légale, de ne pas divulguer cette donnée cruciale.
Malgré l’intention évidente de la FDA d’obscurcir les choses, Pfizer a fourni un moyen d’estimer ce nombre lorsqu’elle a conclu sans équivoque : « … ces analyses de détection de signaux sont cohérentes avec le profil de sécurité connu du vaccin » .
Quel était le profil de sécurité connu du vaccin ?
Au 28 février, le seul profil de sécurité connu du vaccin a été déterminé par les premiers résultats des essais de phase 3 de l’automne 2020.
Sur les 21 621 personnes ayant reçu le vaccin de Pfizer, 126 [Polack FP, Thomas SJ, Kitchin N, et al., NEJM, Tableau S3] ont subi un événement indésirable grave au cours des essais. Cela représente environ un événement indésirable grave sur 171,6 bénéficiaires.
Ainsi, si ces données sont conformes à son profil de sécurité connu, et qu’environ 79 000 événements indésirables graves étaient survenus jusqu’à cette date, nous pouvons estimer qu’environ 13 550 000 (79 000 x 171,6) doses avaient été distribuées.
Certes, il y a une incertitude dans ce calcul. Peut-être une interprétation différente du profil de sécurité était-elle sous-entendue.
Cependant, Pfizer a indiqué le nombre de doses qui avaient été distribuées, et non administrées.
Il y aurait eu moins de doses administrées que de doses délivrées. En outre, les effets indésirables graves observés dans les essais étaient répartis entre les participants qui étaient complètement vaccinés (ayant reçu deux doses).
Ici, nous utilisons le nombre de doses comme dénominateur. Cette estimation permettra d’obtenir la limite inférieure de l’incidence réelle des événements indésirables.
En d’autres termes, en utilisant ces hypothèses, nous accordons au vaccin de Pfizer le plus grand bénéfice du doute.
En utilisant cette estimation des doses totales administrées, l’incidence d’une issue fatale est de 1223/13,55 millions ou 1 sur 11 079.
Séquelles permanentes (affections consécutives à la vaccination) = 520/13,55 millions, soit 1 sur 26 057. En outre, 11 361 sur 13,55 millions, soit 1 sur 1 193, ne s’étaient pas encore remis d’un événement indésirable.
Pfizer a choisi, de manière inexplicable, de regrouper les bénéficiaires qui ont « récupéré » avec ceux qui étaient « en voie de guérison ». Combien de personnes dans ce grand groupe (19 582) souffraient encore d’un préjudice au moment de la rédaction du rapport ? Sur quelle base Pfizer a-t-elle déterminé qu’un receveur avait encore une chance de se rétablir complètement ?
En l’absence d’éclaircissement de la part du fabricant du vaccin, nous sommes contraints de les mettre dans le même panier qu’un autre groupe important de 9 400 personnes dont l’issue finale était « inconnue », ce qui nous laisse une limite élevée de 1 personne sur 466 ayant eu une issue indéterminée.
Bien qu’il ait été démontré qu’aucun de ces événements indésirables et de ces décès n’était directement ou indirectement causé par la vaccination, Pfizer a présenté d’autres données préoccupantes concernant les événements indésirables d’ « intérêt particulier » ( « adverse events of special interest » , AESI).
Selon Pfizer, 1 403 AESI cardiovasculaires, 932 hématologiques, 3 600 musculo-squelettiques, 501 neurologiques et 3 674 « autres » AESI graves sont tous survenus avec un délai médian d’apparition de 24 heures ou moins après la vaccination.
Les 275 accidents vasculaires cérébraux et les 449 cas de paralysie faciale signalés sont survenus avec un délai médian d’apparition de deux jours après la vaccination.
Bien qu’il soit impossible d’établir un lien de causalité inattaquable entre la vaccination et les lésions à l’heure actuelle, la relation temporelle entre les deux est corrélative et hautement suggestive de la causalité.
Néanmoins, les auteurs du rapport de Pfizer ont conclu, à la fin de chaque catégorie de l’AESI, que « cette étude de cas cumulative ne soulève pas de nouveaux problèmes de sécurité » .
Le rapport fait également état de 24 cas graves chez des enfants de moins de 12 ans. Parmi ceux-ci, 13 cas n’avaient pas encore été résolus au moment de la rédaction du rapport. L’âge moyen de ces bénéficiaires était de 3,7 ans.
Nous devons supposer que très peu d’enfants de cet âge ont été inoculés à l’époque, étant donné que Pfizer avait l’autorisation de l’utiliser sur les adultes uniquement. Étant donné que le nombre d’enfants inoculés n’étant pas communiqué, nous ne pouvons pas savoir quel est le risque de blessure chez les enfants de moins de 12 ans.
Conclusions
Les assurances répétées de Pfizer selon lesquelles il n’existe pas de nouveaux problèmes de sécurité sont au mieux fallacieuses.
La FDA a fait ouvertement obstruction en retenant des informations cruciales nécessaires à une évaluation précise des dommages. Cependant, en utilisant des estimations raisonnables basées sur les propres déclarations de Pfizer et les données d’essais publiées, il est probable qu’un signal de sécurité existe – et que ce signal de sécurité a été ignoré par l’organisation même qui est censée l’écouter, la FDA.
L’incidence estimée par Pfizer de la fatalité potentielle du vaccin, 1 sur 11 079, est environ deux fois plus élevée que celle rapportée par le VAERS. Étant donné que les décès potentiels liés aux vaccins mentionnés dans ce document ont fait l’objet d’une déclaration passive, nous pouvons supposer que l’incidence réelle est plus élevée.
Des analyses plus complètes ont démontré un facteur de sous-déclaration de la fatalité des vaccins par le VAERS approchant 41 ou plus.
Qu’elle soit sous-déclarée ou non, la tragédie réelle et croissante est que, tant qu’il n’est pas prouvé qu’une blessure associée à la vaccination est causée par celle-ci, elle reste, à toutes fins utiles, un signal inexistant pour les institutions mêmes responsables de la santé et de la sécurité publiques.
Sur quelles bases pouvons-nous, en tant que médecins et prestataires de soins de santé, assurer à nos patients que ce vaccin est sûr si les effets indésirables ne sont pas étudiés ou même reconnus ?
Le feu vert de la FDA est-il vraiment suffisant ?
Ou devrions-nous exiger la transparence, la discussion ou, à tout le moins, des données non expurgées ? Qu’est-ce que le public attend de nous ?