





En agosto, “Public Health and Medical Professionals for Transparency” (Profesionales de la salud pública y la medicina por la transparencia, PHMPT por sus siglas en inglés) presentó una solicitud de la Ley de Libertad de Información (“Freedom of Information Act “, FOIA por sus siglas en inglés) a la Administración de Alimentos y Medicamentos de los Estados Unidos (2Food and Drug Administration”, FDA por sus siglas en inglés) para obtener todos los datos del expediente del producto biológico de la vacuna COVID-19 de Pfizer, un conjunto de información que comprende unas 329.000 páginas.
La FDA, argumentando que su escaso personal en el Centro de Evaluación e Investigación Biológica no tenía la capacidad de redactar rápidamente el material legalmente exento, como la información de propiedad de Pfizer y la información personal privada de los participantes en el ensayo, la agencia pidió que se le permitiera publicar sólo 500 páginas de estos datos al mes, lo que haría necesarios 55 años para que se llegara a divulgar la información completa.
Posteriormente, la agencia solicitó hasta 75 años para completar la tarea. Hasta el 17 de noviembre, sólo se había publicado una parte de los datos en cuestión.
Aquí analizaré uno de estos documentos publicados, el “Análisis acumulativo de los informes de eventos adversos posteriores a la autorización“. Este documento constituye una parte de la responsabilidad de Pfizer en materia de farmacovigilancia con respecto a su acuerdo de licencia biológica con la FDA.
La farmacovigilancia se refiere a la ciencia y las actividades relacionadas con la detección, evaluación, comprensión y prevención de los efectos adversos o cualquier otro problema relacionado con los medicamentos.
Antes de examinar la cantidad, la gravedad y la naturaleza de los acontecimientos adversos incluidos en este documento, merece la pena detenerse a considerar la importancia que debería tener este informe para el público.
La vacuna de Pfizer aún no había completado todas las pruebas de seguridad y eficacia, pero su producto se estaba desplegando rápidamente en una población sana que empequeñecía el tamaño del ensayo clínico de la vacuna.
La FDA y Pfizer eran muy conscientes de que riesgos muy reales, si es que existían, no podían haber sido identificados sólo con los ensayos. No había suficientes participantes, y los participantes no habían sido observados durante mucho tiempo.
Todo puede parecer bien si se experimenta con 20.000 personas, pero ¿qué ocurre cuando se experimenta con un millón de personas?
El “Análisis acumulativo de los informes de eventos adversos posteriores a la autorización” debería haber proporcionado garantías de que “todo parece estar bien hasta ahora” como buscaba la FDA. ¿Por qué fue necesario impulsar a la FDA a hacer pública esta información mediante una orden judicial?
En la sección de discusión del documento (sección 4), Pfizer asegura a la FDA que “… realiza una detección de señales frecuente y rigurosa en los casos de BNT162b2”.
¿Qué significa la detección “rigurosa” de señales? ¿Pfizer encuestó a un gran número de receptores de vacunas en busca de eventos adversos y los investigó? No, no lo hizo.
Este informe no es más que una recopilación de informes no solicitados, es decir, pasivos, de acontecimientos adversos que los destinatarios han puesto directamente en conocimiento de Pfizer, de casos comunicados por las autoridades sanitarias, de casos publicados en la literatura médica, de casos procedentes de programas de marketing patrocinados por Pfizer, de estudios no intervenidos y de casos de acontecimientos adversos graves comunicados a partir de estudios clínicos, independientemente de la evaluación de la causalidad.
En el informe, Pfizer admitió que “se desconoce la magnitud de la infradeclaración”.
Está bien aceptado que la información pasiva conducirá ineludiblemente a una infradeclaración. Sin embargo, según el informe de Pfizer:
“Debido al gran número de informes de eventos adversos espontáneos recibidos para el producto, el MAH (siglas en inglés de “Marketing Authorisation Holder”, Titular de la Autorización de Comercialización) ha priorizado el procesamiento de los casos graves, con el fin de cumplir con los plazos de notificación reglamentarios acelerados y garantizar que estos informes estén disponibles para la actividad de detección y evaluación de señales.”
Los autores continuaron:
“Pfizer también ha realizado [sic] múltiples acciones para ayudar a aliviar el gran aumento de informes de eventos adversos. Esto incluye importantes mejoras [sic] tecnológicas, y soluciones de procesos y flujos de trabajo, así como el aumento del número de empleados encargados de introducir los datos y del procesamiento de casos.”
En otras palabras, el número de acontecimientos adversos notificados superó las expectativas de Pfizer, aunque el fabricante de la vacuna concluyó: “Los resultados de estos análisis de detección de señales son coherentes con el perfil de seguridad conocido de la vacuna.”
Esta afirmación paradójica será una pista importante cuando analicemos los datos a continuación.
¿Qué revela el documento?
Hasta el 28 de febrero, un total de 42.086 receptores (casos) informaron de 158.893 eventos, o reacciones adversas a la vacuna de Pfizer. Aproximadamente el 50% de estos sucesos se consideraron graves.
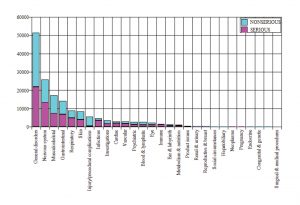
A continuación se ofrece un resumen de las características de los receptores de la vacuna:
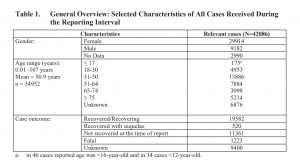
Cabe destacar que 1.223 receptores de la vacuna tuvieron un resultado fatal. Más de 11.000 no se han recuperado. El resultado de 9.400 era desconocido. Casi tres cuartas partes eran mujeres.
Estas cifras son preocupantes, pero ¿representan un problema de seguridad importante? La respuesta a esta pregunta depende totalmente del número de personas que se hayan vacunado hasta ese momento.
Pfizer proporcionó este número a la FDA en la sección de visión general del documento, sección 3.1.1. – pero en el documento publicado en virtud de la solicitud de la FOIA, esa cifra estaba redactada (editada para eliminar u ocultar información que se ha considerado privilegiada o confidencial):
“Se estima que se enviaron aproximadamente (b) (4) dosis de BNT162b2 por todo el mundo desde la recepción de la primera autorización temporal para el suministro de emergencia el 01 de diciembre de 2020 hasta el 28 de febrero de 2021.”
En lo anterior, “(b)(4)” indica que este número ha sido redactado.
El número acumulado de dosis distribuidas en todo el mundo a partir del 28 de febrero no es información altamente sensible, ni incluye datos personales o privados de individuos.
Sin embargo, sin esta cifra clave no hay forma de calcular la incidencia de los acontecimientos graves, es decir, una señal de seguridad.
La FDA optó, sin explicación ni justificación legal alguna, por retener este dato crucial.
A pesar de la intención obvia de la FDA de confundir, Pfizer proporcionó un medio para estimar este número cuando concluyó inequívocamente: “… estos análisis de detección de señales son coherentes con el perfil de seguridad conocido de la vacuna”.
¿Cuál era el perfil de seguridad conocido de la vacuna?
Hasta el 28 de febrero, el único perfil de seguridad conocido de la vacuna estaba determinado por los resultados iniciales de los ensayos de fase 3 del otoño de 2020.
De los 21.621 receptores de la vacuna de Pfizer, 126[Polack FP, Thomas SJ, Kitchin N, et al., NEJM, Tabla S3] sufrieron un acontecimiento adverso grave en los ensayos. Esto supone aproximadamente un acontecimiento adverso grave por cada 171,6 receptores.
Por tanto, si estos datos son coherentes con su perfil de seguridad conocido, y hasta ese momento se habían producido aproximadamente 79.000 acontecimientos adversos graves, podemos estimar que se habían distribuido aproximadamente 13.550.000 (79.000 x 171,6) dosis.
Ciertamente hay incertidumbre en este cálculo. Tal vez se haya dado a entender una interpretación diferente del perfil de seguridad.
Sin embargo, Pfizer informó del número de dosis que se habían distribuido, no de las administradas.
Se habrían administrado menos dosis de las que se entregaron. Además, los acontecimientos adversos graves en los ensayos se distribuyeron entre los participantes que estaban totalmente vacunados (que habían recibido dos dosis).
Aquí utilizamos el número de dosis como denominador. Esta estimación dará como resultado el límite inferior de la verdadera incidencia de eventos adversos.
En otras palabras, al utilizar estos supuestos estamos dando a la vacuna de Pfizer el máximo beneficio de la duda.
Utilizando esta estimación del total de dosis administradas, la incidencia de un resultado mortal es de 1223/13,55 millones o 1 de cada 11.079.
Secuelas permanentes (afecciones derivadas de la vacunación) = 520/13,55 millones, es decir, 1 de cada 26.057. Además, 11.361 de 13,55 millones, es decir, 1 de cada 1.193, aún no se había recuperado de un evento adverso.
Pfizer eligió inexplicablemente agrupar a los receptores que se “recuperaron” con los que estaban “recuperándose”. ¿Cuántos de este gran grupo (19.582) seguían sufriendo daños en el momento del informe? ¿En qué se basó Pfizer para determinar que un receptor aún tenía posibilidades de recuperarse por completo?
Al no haber ninguna aclaración por parte del fabricante de la vacuna, nos vemos obligados a agruparlos con otro gran grupo de 9.400 cuyo resultado final era “desconocido”, lo que nos deja con un límite alto de 1 de cada 466 receptores que han tenido un resultado indeterminado.
Aunque no se demostró que ninguno de estos acontecimientos adversos y muertes fueran causados directa o indirectamente por la vacunación, Pfizer ofreció más datos preocupantes en torno a los acontecimientos adversos de “especial interés” (“adverse events of special Interest”, AESI por sus siglas en inglés).
Según Pfizer, se produjeron 1.403 IAES cardiovasculares, 932 hematológicas, 3.600 musculoesqueléticas, 501 neurológicas y 3.674 “otras” AESI graves con una mediana de tiempo de aparición de 24 horas o menos desde la vacunación.
Los 275 accidentes cerebrovasculares y los 449 casos de parálisis facial notificados se produjeron con una media de tiempo de aparición de dos días desde la vacunación.
Aunque en este momento es imposible establecer una relación causal indiscutible entre la vacunación y las lesiones, la relación temporal entre ellas es correlativa y altamente sugerente de causalidad.
Sin embargo, los autores del informe de Pfizer concluyeron al final de cada categoría de AESI que “Esta revisión de casos acumulados no plantea nuevos problemas de seguridad.”
El informe también incluía 24 casos graves en niños menores de 12 años. De ellos, 13 casos aún no se habían resuelto en el momento de redactar el informe. La edad media de estos receptores era de 3,7 años.
Debemos suponer que muy pocos niños de esa edad fueron inoculados en esa época, dado que Pfizer tenía autorización para su uso sólo en adultos. Al no haberse comunicado el número de niños inoculados, no podemos saber cuál es el riesgo de lesión en los menores de 12 años.
Conclusiones
Las repetidas garantías de Pfizer de que no existen nuevos problemas de seguridad son, en el mejor de los casos, poco sinceras.
La FDA se mostró abiertamente obstruccionista al ocultar información crucial necesaria para realizar una evaluación precisa de los daños. Sin embargo, utilizando estimaciones razonables basadas en las propias afirmaciones de Pfizer y en los datos de los ensayos publicados, es probable que exista una señal de seguridad – y que esa señal de seguridad haya sido ignorada por la misma organización que se supone que debería estar atenta a ella, la FDA.
La incidencia estimada por Pfizer de una posible letalidad de la vacuna, 1 de cada 11.079, es aproximadamente el doble de la notificada en el VAERS. Dado que las posibles víctimas mortales de las vacunas en este documento se han notificado de forma pasiva, podemos suponer que la incidencia real es mayor.
Análisis más exhaustivos han demostrado que el factor de subnotificación del VAERS en cuanto a la letalidad de las vacunas se acerca al 41 o más.
Con o sin información, la tragedia real y creciente es que hasta que no se demuestre que una lesión asociada a la vacunación es causada por ella, sigue siendo, a todos los efectos, una señal inexistente para las propias instituciones responsables de la salud y la seguridad públicas.
¿En qué podemos basarnos los médicos y los profesionales sanitarios para asegurar a nuestros pacientes que esta vacuna es segura si no se investigan los efectos adversos o ni siquiera se reconocen?
¿Es suficiente un visto bueno de la FDA?
¿O debemos exigir transparencia, debate o, al menos, datos no redactados? ¿Qué espera el público de nosotros?