





Im August reichte die Public Health and Medical Professionals for Transparency (PHMPT) einen Antrag auf Auskunftserteilung unter dem Freedom of Information Act (FOIA) bei der US-amerikanischen Food and Drug Administration (FDA) ein, um alle Daten in der biologischen Produktakte des COVID-19-Impfstoffs von Pfizer zu erhalten – ein Informationspaket, das etwa 329.000 Seiten umfasst.
Die FDA argumentiert, dass das personell schlecht ausgestattete Center for Biologics Evaluation and Research nicht in der Lage war, rechtlich geschütztes Material, wie z. B. geschützte Informationen von Pfizer und persönliche Informationen von Studienteilnehmern, schnell auszusortieren. Die Agentur bat daher darum, nur 500 Seiten dieser Daten pro Monat freigeben zu müssen. 55 Jahre für die volle Offenlegung.
Später beantragte die Agentur bis zu 75 Jahre, um der Anfrage nachzukommen. Bis zum 17. November war nur ein Bruchteil der fraglichen Daten veröffentlicht worden.
Im Folgenden werde ich eines dieser freigegebenen Dokumente besprechen, die “Cumulative Analysis of Post-authorization Adverse Event Reports” (dt.: “Kumulative Analyse der Berichte über unerwünschte Ereignisse nach der Zulassung”). Dieses Dokument stellt einen Teil der Verantwortung von Pfizer für die Pharmakovigilanz in Bezug auf ihren Biologielizenzvertrag mit der FDA dar.
Der Begriff Pharmakovigilanz bezieht sich auf die Wissenschaft und die Tätigkeiten im Zusammenhang mit der Erkennung, der Bewertung, dem Verständnis und der Vorbeugung von unerwünschten Nebenwirkungen oder anderen Problemen im Zusammenhang mit Arzneimitteln.
Bevor wir uns mit der Anzahl, der Schwere und der Art der in diesem Dokument enthaltenen unerwünschten Ereignisse befassen, lohnt es sich, innezuhalten und zu überlegen, wie bedeutsam dieser Bericht für die Öffentlichkeit hätte sein können.
Der Impfstoff von Pfizer hatte noch keine vollständigen Sicherheits- und Wirksamkeitstests durchlaufen, doch sein Produkt wurde schnell in einer gesunden Bevölkerung eingesetzt, die die Größe der klinischen Studie des Impfstoffs in den Schatten stellte.
Die FDA und Pfizer waren sich sehr wohl bewusst, dass sehr reale Risiken, sofern sie existierten, durch die Versuche allein nicht hätten erkannt werden können. Es gab nicht genügend Teilnehmer, und die Teilnehmer wurden nicht sehr lange beobachtet.
Wenn man an 20.000 Menschen experimentiert, mag alles in Ordnung sein, aber was passiert, wenn man an einer Million Menschen experimentiert?
Die “Kumulative Analyse der Berichte über unerwünschte Ereignisse nach der Zulassung” hätte der FDA die Gewissheit geben sollen, dass “bisher alles gut aussieht”. Warum war es notwendig, die FDA durch einen Gerichtsbeschluss zu zwingen, diese Informationen zu veröffentlichen?
Im Diskussionsteil des Dokuments (Abschnitt 4) versichert Pfizer der FDA, dass es “… eine häufige und strenge Signalerkennung bei BNT162b2-Fällen durchführt”.
Was bedeutet “rigorose” Signalerkennung? Hat Pfizer eine große Anzahl von Impfstoffempfängern nach unerwünschten Ereignissen befragt und diese untersucht? Nein, das hat sie nicht.
Bei diesem Bericht handelt es sich lediglich um eine Zusammenstellung von unaufgeforderten, d. h. passiven Berichten über unerwünschte Ereignisse, die Pfizer direkt von den Empfängern zur Kenntnis gebracht wurden, von Fällen, die von den Gesundheitsbehörden gemeldet wurden, von Fällen, die in der medizinischen Fachliteratur veröffentlicht wurden, von Fällen aus von Pfizer gesponserten Marketingprogrammen, von nicht-interventionellen Studien und von Fällen schwerwiegender unerwünschter Ereignisse, die aus klinischen Studien unabhängig von der Bewertung der Kausalität gemeldet wurden.
In dem Bericht räumte Pfizer ein, dass das “Ausmaß der Untererfassung unbekannt ist”.
Es ist allgemein anerkannt, dass eine passive Berichterstattung unweigerlich zu einer Untererfassung führt. Dem Bericht von Pfizer zufolge ist dies jedoch nicht der Fall:
“Aufgrund der großen Anzahl von Spontanmeldungen von unerwünschten Ereignissen für das Produkt hat der Zulassungsinhaber (MAH) die Bearbeitung von schwerwiegenden Fällen priorisiert, um die beschleunigten Meldefristen der Behörden einzuhalten und sicherzustellen, dass diese Meldungen für die Signalerkennung und -bewertung zur Verfügung stehen.”
Die Autoren fuhren fort:
“Pfizer hat außerdem unter [sic] mehrere Maßnahmen ergriffen, um den starken Anstieg der Berichte über unerwünschte Ereignisse zu lindern. Dazu gehören erhebliche technologische Verbesserungen, [sic], Prozess- und Workflow-Lösungen sowie die Aufstockung der Zahl der Mitarbeiter in der Dateneingabe und Fallbearbeitung.”
Mit anderen Worten: Die Zahl der gemeldeten unerwünschten Ereignisse übertraf die Erwartungen von Pfizer, und dennoch kam der Impfstoffhersteller zu dem Schluss: “Die Ergebnisse dieser Signaldetektionsanalysen stimmen mit dem bekannten Sicherheitsprofil des Impfstoffs überein.”
Diese paradoxe Aussage wird sich als wichtiger Hinweis erweisen, wenn wir die Daten weiter unten auswerten.
Was geht aus dem Dokument hervor?
Bis zum 28. Februar meldeten insgesamt 42.086 Empfänger (Fälle) 158.893 Ereignisse oder unerwünschte Reaktionen auf den Impfstoff von Pfizer. Ungefähr 50 % dieser Ereignisse wurden als schwerwiegend eingestuft.
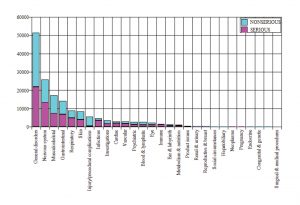
Hier finden Sie einen Überblick über die Eigenschaften der Empfänger:
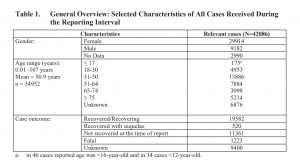
Bemerkenswert ist, dass 1.223 Empfänger des Impfstoffs einen tödlichen Ausgang hatten. Mehr als 11.000 hatten sich nicht erholt. Das Ergebnis von 9.400 war unbekannt. Nahezu drei Viertel der Befragten waren weiblich.
Diese Zahlen sind besorgniserregend, aber stellen sie auch ein erhebliches Sicherheitsrisiko dar? Die Antwort auf diese Frage hängt ganz davon ab, wie viele Menschen bis zu diesem Zeitpunkt geimpft worden sind.
Pfizer hat der FDA diese Zahl im allgemeinen Überblick des Dokuments, Abschnitt 3.1.1. mitgeteilt, – aber in dem auf Grundlage des FOIA-Antrags freigegebenen Dokument wurde diese Zahl geschwärzt:
“Es wird geschätzt, dass etwa (b) (4) Dosen von BNT162b2 seit dem Erhalt der ersten befristeten Genehmigung für die Notfallversorgung am 01. Dezember 2020 bis zum 28. Februar 2021 weltweit versandt wurden.”
Im obigen Text bedeutet “(b)(4)”, dass diese Nummer geschwärzt wurde.
Die kumulative Anzahl der bis zum 28. Februar weltweit verteilten Dosen ist keine geschützte Information und stellt auch keine persönlichen, privaten Daten von Einzelpersonen dar.
Doch ohne diese Schlüsselzahl gibt es keine Möglichkeit, die Inzidenz schwerwiegender Ereignisse, d. h. ein Sicherheitssignal, zu berechnen.
Die FDA hat sich ohne Erklärung oder rechtliche Begründung dafür entschieden, diese wichtigen Daten zurückzuhalten.
Trotz der offensichtlichen Absicht der FDA zu verschleiern, lieferte Pfizer ein Mittel zur Schätzung dieser Zahl, als es unmissverständlich zu dem Schluss kam: “… diese Analysen zur Signalerkennung sind mit dem bekannten Sicherheitsprofil des Impfstoffs vereinbar.”
Welches Sicherheitsprofil des Impfstoffs war bekannt?
Bis zum 28. Februar war das einzige bekannte Sicherheitsprofil des Impfstoffs durch die ersten Ergebnisse der Phase-3-Studien vom Herbst 2020 bestimmt.
Von 21.621 Empfängern des Impfstoffs von Pfizer erlitten 126[Polack FP, Thomas SJ, Kitchin N, et al., NEJM, Tabelle S3] in den Studien ein schwerwiegendes unerwünschtes Ereignis. Dies entspricht ungefähr einem schweren unerwünschten Ereignis bei 171,6 Empfängern.
Wenn diese Daten mit dem bekannten Sicherheitsprofil übereinstimmen und bis zu diesem Zeitpunkt etwa 79.000 schwerwiegende unerwünschte Ereignisse aufgetreten sind, können wir schätzen, dass etwa 13.550.000 (79.000 x 171,6) Dosen verteilt worden sind.
Zugegebenermaßen ist diese Berechnung mit Unsicherheiten behaftet. Vielleicht wurde das Sicherheitsprofil anders interpretiert.
Pfizer gab jedoch die Anzahl der verteilten und nicht der verabreichten Dosen an.
Es wären weniger Dosen verabreicht als ausgeliefert worden. Außerdem verteilten sich die schwerwiegenden unerwünschten Ereignisse in den Studien auf Teilnehmer, die vollständig geimpft waren (zwei Dosen erhalten hatten).
Hier wird die Anzahl der Dosen als Nenner verwendet. Diese Schätzung ergibt die untere Grenze der tatsächlichen Inzidenz von unerwünschten Ereignissen.
Mit anderen Worten: Mit diesen Annahmen geben wir dem Impfstoff von Pfizer den größtmöglichen Vertrauensvorschuss.
Ausgehend von dieser Schätzung der insgesamt verabreichten Dosen beträgt die Inzidenz eines tödlichen Ausgangs 1.223/13,55 Millionen oder 1 von 11.079.
Dauerhafte Folgeerkrankungen (Erkrankungen als Folge der Impfung) = 520/13,55 Millionen, d. h. 1 von 26.057. Darüber hinaus hatten sich 11.361 von 13,55 Millionen, also 1 von 1.193, noch nicht von einem Zwischenfall erholt.
Pfizer hat sich unerklärlicherweise dafür entschieden, die Empfänger, die “genesen” sind, mit denjenigen zu gruppieren, die “sich erholen”. Wie viele aus dieser großen Gruppe (19.582) waren zum Zeitpunkt des Berichts noch geschädigt? Auf welcher Grundlage hat Pfizer festgestellt, dass ein Empfänger noch eine Chance auf vollständige Genesung hat?
Da der Impfstoffhersteller keine Angaben gemacht hat, sind wir gezwungen, sie mit einer anderen großen Gruppe von 9.400 Personen zusammenzulegen, deren Endergebnis “unbekannt” war, so dass wir von einer hohen Zahl von 1 von 466 Empfängern mit unbestimmtem Ergebnis ausgehen.
Obwohl keine dieser unerwünschten Ereignisse und Todesfälle nachweislich direkt oder indirekt durch die Impfung verursacht wurden, legte Pfizer weitere besorgniserregende Daten zu unerwünschten Ereignissen von “besonderem Interesse” (AESI) vor.
Nach Angaben von Pfizer traten 1.403 kardiovaskuläre AESIs, 932 hämatologische, 3.600 muskuloskelettale, 501 neurologische und 3.674 “andere” schwerwiegende AESIs auf, die im Median 24 Stunden oder weniger nach der Impfung auftraten.
Die 275 gemeldeten Schlaganfälle und 449 Fälle von Gesichtslähmung traten im Median zwei Tage nach der Impfung auf.
Obwohl es unmöglich ist, zum jetzigen Zeitpunkt einen eindeutigen kausalen Zusammenhang zwischen Impfung und Schädigung herzustellen, ist der zeitliche Zusammenhang zwischen beiden korrelativ und deutet stark auf eine Kausalität hin.
Dennoch kamen die Autoren des Pfizer-Berichts am Ende jeder AESI-Kategorie zu dem Schluss, dass “diese kumulative Fallprüfung keine neuen Sicherheitsprobleme aufwirft”.
Der Bericht umfasste auch 24 schwere Fälle bei Kindern unter 12 Jahren. Davon waren 13 Fälle zum Zeitpunkt der Berichterstattung noch nicht abgeschlossen. Das Durchschnittsalter dieser Empfänger betrug 3,7 Jahre.
Wir müssen davon ausgehen, dass damals nur sehr wenige Kinder in diesem Alter geimpft worden waren, da Pfizer nur eine Zulassung für Erwachsene hatte. Da keine Angaben über die Zahl der geimpften Kinder vorliegen, können wir nicht wissen, wie hoch das Verletzungsrisiko bei Kindern unter 12 Jahren ist.
Schlussfolgerungen
Die wiederholten Zusicherungen von Pfizer, dass keine neuen Sicherheitsprobleme bestehen, sind bestenfalls unaufrichtig.
Die FDA verhielt sich offenkundig hinderlich, indem sie entscheidende Informationen zurückhielt, die für eine genaue Bewertung der Schäden erforderlich sind. Bei Verwendung vernünftiger Schätzungen auf der Grundlage der eigenen Angaben von Pfizer und der veröffentlichten Studiendaten ist es jedoch wahrscheinlich, dass es ein Sicherheitssignal gibt – und dass dieses Sicherheitssignal von genau der Organisation ignoriert wurde, die eigentlich darauf achten sollte, nämlich der FDA.
Die von Pfizer geschätzte Inzidenz potenzieller Impftodesfälle, 1 zu 11.079, ist etwa doppelt so hoch wie die von VAERS gemeldete. Da die in diesem Dokument genannten potenziellen Impftodesfälle passiv gemeldet wurden, können wir davon ausgehen, dass die tatsächliche Inzidenz höher ist.
Umfassendere Analysen haben gezeigt, dass die Zahl der von VAERS gemeldeten Impftodesfälle bei 41 oder mehr liegt.
Ob zu wenig berichtet wird oder nicht, die wirkliche und wachsende Tragödie besteht darin, dass ein Schaden, der mit einer Impfung in Verbindung gebracht wird, bis zu dem Zeitpunkt, an dem nachgewiesen wird, dass er durch die Impfung verursacht wurde, in jeder Hinsicht ein nicht existierendes Signal für die Institutionen ist, die für die öffentliche Gesundheit und Sicherheit verantwortlich sind.
Auf welcher Grundlage können wir als Ärzte und Gesundheitsdienstleister unseren Patienten versichern, dass dieser Impfstoff sicher ist, wenn unerwünschte Ereignisse nicht untersucht oder sogar anerkannt werden?
Ist eine Zustimmung der FDA wirklich gut genug?
Oder sollten wir Transparenz, Diskussion oder zumindest ungeschwärzte Daten fordern? Was erwartet die Öffentlichkeit von uns?